
The best way to prevent and remove infections is through the natural ‘sterilising’ action of the immune response that combines elements of both innate and adaptive immunity to ward off foreign pathogens without medical intervention. The immune system ‘remembers’ the cleared foreign antigens to speed up its response to re-infection. The immune system in most cancer patients can still completely destroy viruses and bacteria. The ferocity and specificity of this response can be witnessed in the way an inadequately suppressed immune system can completely destroy a large transplanted organ, while sparing one’s own (self) tissues. This destructive effect would be beneficial for cancer therapy if it could be directed at tumours.
The field of immunotherapy seeks methods to harness, direct and control this immunity especially against cancer. Therapeutic cancer vaccines are a form of immunotherapy designed to educate the immune system of patients with existing cancers to recognise their tumour cells as foreign rather than self. If successful, an immune response could theoretically stimulate immune cells to destroy large tumours, and also seek out and destroy metastatic tumour cells. After immunotherapy, the ability of the immune system to ‘remember’ tumour cells ought to eliminate any recurrence without additional treatment in much the same way as it protects against opportunistic infections.
The immune system promises complete tumour elimination and durable remissions with properly designed cancer vaccines
Immunotherapy is a highly desirable alternative (or complement) to current treatment strategies. Unlike immune-mediated anti-tumour mechanisms, surgery, radiation and chemotherapy have no anti-tumour specificity at the single cell level. Therefore, it is not technologically feasible for them to eliminate every last tumour cell, without which cancer recurrence commonly occurs. Furthermore, these modalities lead to resistance rather than ‘memory’ of tumour.
The immune system promises complete tumour elimination and durable remissions with properly designed cancer vaccines. Its specificity in targeting malignant cells and avoiding normal cells also promises minimal toxicity. This is the only currently feasible approach that has the technical possibility to cure cancer.
Current immunotherapy development status
The ability to educate the immune system to remove all cancer cells has been successful in some animal models. Mice can be immunised to reject syngeneic transplanted tumours and protect against tumour rechallenge without further treatment, whereas nonimmunised mice invariably die. Protection is tumourspecific, since immunised mice succumb to challenges from different tumours. Tumour-specific CD8+ cytotoxic T-cells (CTL) are mainly responsible for tumour elimination. Moreover, CTLs can specifically recognise tumour cells and do not attack normal cells of the same tissue.
While these animal studies provide “proof-ofconcept” for translating these methods to the clinic, the promise of immunotherapy has been difficult to realise. In the last two decades, immunological strategies have been designed to stimulate antitumour immunity in cancer patients, by vaccination with peptide-pulsed or tumour lysate-pulsed dendritic cells, cytokine-mediated immunotherapy, the administration of large numbers of T cells generated from tumour-infiltrating lymphocytes or engineered to express receptors for specific tumour-associated antigens (TAA), ‘naked DNA’, and recombinant virus. Despite numerous attempts, the success rate of clinical immunotherapy remains abysmally low. However encouraging reports of successful trials using novel cancer vaccines in prolonging survival in prostate cancer illustrates the potential of this approach, although the results fall far short of the curative potential.
Despite the poor clinical results, dozens of cancer vaccine clinical trials are currently being conducted by both industrial and academic sponsors. One of the reasons for continuing development and testing of anti-cancer vaccines is the demand for alternatives to the high morbidity associated with other modalities of treatment, since immunotherapy has little toxicity. While response rates to highly toxic chemotherapy have improved over the last two decades, the modest increase in 5-year survival [1] has come at a severe price in terms of quality of life.
Flaws in cancer vaccine development
The failure of hundreds of immunotherapy clinical trials to produce significant anti-tumour activity calls for a re-examination of the underlying principles. For over 200 years, vaccines have been used successfully to prevent numerous infectious diseases by inducing a humoral response (antibodies). The same concept is being applied to develop cancer vaccines with the intention of treating existing tumours, despite the fact that vaccines giving protection against pathogenic infection are incapable of curing existing infections with the same pathogen. Hence, modeling of cancer therapy on the basis of protective immunity is fundamentally flawed as a strategy because protective immunity does not provide therapeutic immunity. Therapeutic immunity to cancer requires a cellular immune response.
Cancer vaccination has followed classical development strategies by focusing on finding unique antigens on tumours not found on normal cells, i.e. tumour-specific antigens (TSA), or by seeking tumourassociated antigens (TAA) overexpressed by cancer cells. TSAs have only been found in tumours induced by infectious agents (e.g. EBNA-1 antigen from Epstein Barr virus-induced Burkitt’s lymphoma). However, many TAAs have been identified, including CEA, MUC1, HER-2/neu and α-fetal protein, of which some are expressed on many tumour types, but not on normal tissues, with the exception of spermatogonia (e.g. MAGE family, GAGE family and NYESO- 1 antigens). TAAs are self antigens and thus do not mark tumours as foreign, but nevertheless enable immunological distinction between tumour and normal cells.
Cancer vaccines containing TAA can also contain agents that augment the ability of these antigens to stimulate anti-tumour immune responses. These have included mixture with immunological adjuvants (such as MF59, incomplete Freund’s adjuvant, saponins QS-21, and bacillus Calmette-Guerin [BCG]), synthesis of more immunogenic derivatives, conjugation to immunogenic proteins, and pulsing directly to dendritic cells, but without much success.
The perceived need for ‘augmenting’ TAA is based on the assumption that, since TAAs are derived from self tissues, rather than from foreign pathogens as in previous vaccines, they are relatively weak. Methods that enhance the immunogenicity of TAA are thought to be essential for eliciting stronger responses that can have a clinical anti-tumour effect. The assumption is that cancer is a disease of a weakened immune system and, therefore, methods are required to ‘boost’ the immune system to treat cancer. Traditional immunology has taught us that the main purpose of the immune system is to distinguish between “self” and “non-self”, and since cancer is “self”, there should be no immune response against it, a concept which is also fundamentally flawed.
Cancer: a disease of a weak immune system?
When a normal cell transforms into a tumour cell, changes occurring in the surface expression of antigens give rise to TAAs that theoretically can be detected by immune cells. Ehrlich first posited in 1909 that the immune system protects the host against cancer, a concept modified in the 1950s by Lewis Thomas and later by Nobel laureate Sir Macfarlane Burnet, who proposed that the immune system has a “surveillance” mechanism for eliminating precancerous and cancerous cells. The fundamental tenet of this hypothesis is that tumours arise constantly in the body and the immune system must recognise and eliminate cells that express TAAs. It predicts that in circumstances where the immune system is weaken or suppressed, surveillance is compromised, tumour cells take hold and clinical disease results. This has been the basis on which the immune system in cancer cases is seen as weak, and so the immune system must be strengthened to mount an attack against cancer. However, there is much evidence to the contrary.
While a few rare cancers occur in immunosuppressed individuals, most human cancers form in immunocompetent individuals. Additionally, there is no doubt that many neoplasms, particularly those of epithelial origin, have a significant inflammatory cell component. This includes a diverse leukocyte infiltrate of macrophages, neutrophils, eosinophils, and mast cells, often in association with lymphocytes. Tumours are usually abundantly infiltrated with immune cells, which argues against weak recognition of their antigens. Further, many attempts to augment the immune response enhance rather than suppress tumour growth [2,3]. In mice, a newly induced in situ tumour, both of mesenchymal and epithelial derivation, can be stimulated to grow faster if it engenders an immune response. Even highly immunogenic tumours left undisturbed in their original hosts can grow faster than tumours with little or no immunogenicity. Therefore, the idea that tumours are “invisible” to the immune system, which therefore needs to be boosted, is misconceived. The problem may be that the immune response to the tumour, while strong, may be of the wrong type. In attempting to boost an immune response that has already failed to protect against tumour formation, it is not surprising that tumour growth may be enhanced rather than suppressed. This is another flaw in the strategy of cancer vaccine development.
Right versus wrong immune response to cancer
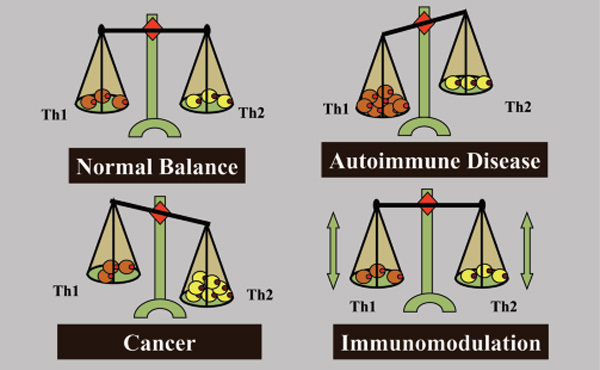
Before designing a cancer vaccine of therapeutic potential, the type of immunity that is required for tumour elimination needs to be understood. Immune responses are generally described by two polarised responses, the T-helper type 1 (Th1) and the T-helper type 2 (Th2). A Th1 response mediates cellular immunity and is critical for immune-mediated tumour eradication; a Th2 response mediates humoral immunity, the type of immunity that can protect against some infectious diseases, but is the “wrong” or inappropriate response to a tumour. Tumour-mediated deviation of T-helper cell differentiation to Th2 is a tumour strategy for immunoavoidance and survival. In fact, an ineffective Th2 response can be detected in most patients with advanced cancer and metastatic disease. This explains why simply boosting the immune response in cancer patients fails, as this only serves to enhance a resident Th2 response that has already failed to protect against tumour formation and is incapable of eradicating cancer.
Th1 and Th2 responses are counter-regulatory, increased Th1 responses downregulate Th2 responses and vice versa. Therefore, we propose that one function of a successful cancer vaccine candidate would be to avoid enhancing an existing “wrong” Th2 response, and to create de novo an excessive Th1 “right” response to the tumour sufficient to downregulate the Th2 response. The challenge will be to create this Th1 response against an overwhelming tumourmediated resident Th2 response and natural mechanisms that suppress autoimmune destruction of normal tissues. However, deviation of the anti-tumour immune response from a Th2 to a Th1 response can only be a part of a possible strategy for therapeutic anti-tumour immunity. While Th1 immunity is essential for tumour eradication, in some patients who develop a potentially effective Th1 response against tumours (or can be immunised to develop one), their tumours continued to grow. This shows that tumours can successfully suppress or avoid a protective Th1 response. Accordingly, a successful immunotherapy strategy will not only need to enhance Th1 immunity and suppress Th2 immunity, but must disable immunoavoidance of the Th1 response and counter natural tolerance circuits preventing immune destruction of normal tissues.
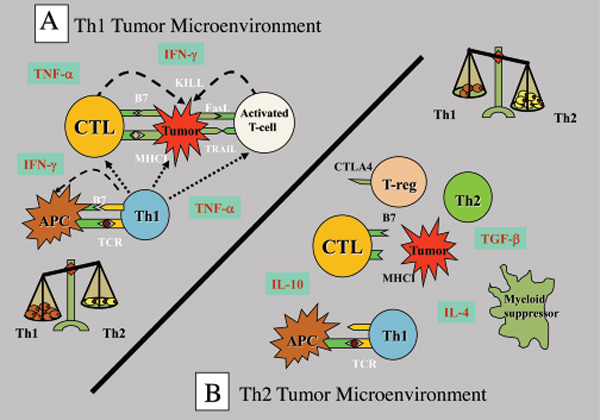
Tumour immunoavoidance

Tumours have numerous active mechanisms to suppress host Th1 immunity, including altering the function of APCs, fostering dysfunctional T-cell co-signaling, generating an immune-subversive Th2 cytokine milieu, downregulating HLA molecules, and recruiting tolerogenic DCs and myeloid suppressor cells. Many of the mechanisms that impede anti-tumour immunity result in the development of T regulatory (Treg) cells. These Treg cells are the dominant immune escape mechanism in early tumour progression. In addition, most human tumour cells produce high levels of transforming growth factor (TGF)-β that can directly convert naïve T cells to Treg cells. TGF-β also suppresses the transcription of genes encoding multiple key proteins of CD8+ cytotoxic T-cells (CTL), such as the cytolytic molecules perforin and granzymes.
Tumour-mediated immunoavoidance shifts and promotes the immune system to an ineffective Th2 response which renders it tolerant, permitting tumours to grow unimpeded by the surveillance mechanisms. Establishment of self-tolerance is a part of a natural immune regulatory mechanism that prevents autoimmune reactions to organ-specific self-antigens. Many such tolerance mechanisms are the same as those employed by tumours to prevent immune destruction.
Therefore, an additional feature of a successful therapeutic cancer vaccine is the ability to overcome cancer immunoavoidance mechanisms and natural tolerance mechanisms.
Strategy to overcome cancer immunoavoidance
Since the immune system is programmed to respond aggressively to foreign antigens, it has perplexed immunologists as to how a successful pregnancy is possible. The fetus is foreign to the mother (50% match) and mother’s immune cells regularly encounter foreign cells of the fetus, yet they do not respond these cells. The interaction between the tumour and the immune system has been likened to pregnancy where an allogeneic graft (the fetus) rapidly develops without rejection by an immunologically competent host. The ability of the fetus to evade the maternal immune response is not due to anatomical barriers, since maternal immune cells cross the placenta and can enter the fetus. The mechanisms by which tumours escape immune attack are similar to the escape of the allogeneic fetoplacental unit, which include loss or downregulation of HLA molecules, Th2 cytokine activity shift, secretion of immunosuppressive factors (including TGF-β), Treg activation, and suppression of Th1 immunity.
Tolerance to a fetus is lost in some women experiencing spontaneous abortion. The mechanism of breakdown of natural immune protection may, therefore, be instructive to cancer vaccine design. There is a unique cytokine network at the materno-fetal interface that plays a key role in the maintenance of pregnancy. Successful pregnancy is correlated with a Th2 bias both at the materno-fetal interface and in the periphery. However, in an abortionprone pregnancy, a Th1 bias occurs systemically and at the maternofetal interface. Like the materno-fetal interface, ongoing inflammatory responses within the stroma of tumours are also dominated by Th2 cytokines that not only promote tumour growth, but profoundly suppress the host’s ability to mount an effective Th1 anti-tumour immune response. This suggests that conversion of the systemic and intratumour microenvironment to Th1 may break immune tolerance to the tumour.
Evidence in support of this concept can be found by analyzing the potent anti-tumour immune effect that occurs in allogeneic bone marrow/stem cell transplantation (BMT) setting. The graft versus tumour (GVT) response is arguably the most potent form of cancer immunotherapy in clinical use. In fact, the GVT effect that occurs on infusion of donor lymphocytes in CML patients with a tumour recurrence after allogeneic BMT is the only way to cure the disease.
Clinical application of GVT is limited, however, due to graft versus host disease (GVHD), a life-threatening complication of allogeneic BMT. T-cells in the donor graft are critical in the induction of both GVHD and GVT because depletion of T-cells from the bone marrow graft not only effectively prevents GVHD, but results in more frequent cancer relapse. Both GVT and GVHD are mediated by production of Th1 cytokines, making it difficult to separate the beneficial GVT effects from the harmful GVHD effects.
The anti-tumour GVT effect provides long-term remission when it occurs through the development of Th1 tumour-specific immunity. This suggests that GVT is not simply the result of tumour eradication by allogeneic recognition. Immunity that protects against tumour recurrence without further treatment suggests development of tumour-specific adaptive immunity. Indeed, donor-derived tumourspecific CTL develops in the host and contributes to the GVT effect. Thus the sustained Th1 cytokine release that occurs in allogeneic BMT due to GVHD acts as an adjuvant in creating anti-Th1 immunity, breaking immune tolerance, and disabling tumour immune suppression that can result in durable remission from disease.
Understanding human immune mechanisms involved in normal pregnancy and spontaneous abortion provides a model for designing vaccine immune responses to overcome self-tolerance
In the past, immunotherapy methods have been translated from animal models to the clinic, but without significant anti-tumour efficacy. Understanding human immune mechanisms involved in normal pregnancy and spontaneous abortion provides a model for designing vaccine immune responses to overcome self-tolerance. The GVT effect of allogeneic BMT shows how Th1 anti-tumour immunity might develop in a cancer patient de novo and possibly overcome tumour immunoavoidance.
New ideas on the development of cancer vaccines

This discussion provides a working hypothesis that can explain the failures of immunotherapy in the clinic. Past immunotherapy approaches have relied upon proof-of-concept from animal data that has not translated well to the clinic; classic methods of vaccine development which provide protective rather than therapeutic immunity, and which either acted to stimulate a resident ineffective Th2 response or promoted a Th1 anti-tumour response without disabling tumour immunoavoidance and natural tolerance mechanisms.
Successful immunotherapy will require the conversion of a resident Th2 immune response into a Th1 response. De novo development of a Th1 response that overwhelms and disables the resident Th2 response is feasible if antigens are introduced in the presence of Th1 cytokines, as occurs in the curative GVT effect after allogeneic BMT. The presence of Th1 cytokines can drive uncommitted T-cells to develop a Th1 cytokine profile, while simultaneously inhibiting cells with the reciprocal phenotype; e.g. IFN-γ, a Th1 cytokine, can selectively expand Th1 cells and inhibit proliferation of Th2 cells. Further, breaking of tolerance to autologous tissues is also feasible in a sustained Th1 inflammatory environment, as evidenced by Th1-mediated spontaneous abortion and Th1-mediated GVHD, which acts as an adjuvant to the curative Th1 anti-tumour effect in allogeneic BMT procedures.
These mechanisms are consistent with the new concept called the “danger theory”, which proposes that the regulation of immunity and tolerance is not determined by recognition of self versus foreign antigens as previously thought, but by the context in which the antigens are presented to the immune system [4,5]. Thus antigen encounter by dendritic cells (DC) in an inflammatory or ‘dangerous’ environmental context prompts a strong Th1 immune response regardless of whether the antigens are self- or foreign-derived. In contrast, antigen capture by DC in a non-inflammatory context leads ultimately to immunotolerance. A tumour “conditions” the microenvironment for the tolerant response, but where normal cells are being destroyed as in GVHD, an active Th1 response is favoured.
A new experimental cancer vaccine protocol
To elicit anti-tumour immunity, it is necessary to engineer the microenvironment where DCs are processing tumour antigens to contain sufficient inflammatory “danger signals” potent enough to downregulate tumour-mediated immunosuppressive cytokine production and related tolerogenic mechanisms. This should enable the development of a Th1 immune response against the tumour, whose magnitude and duration will also control whether it is sufficient to downregulate tumour immunoavoidance mechanisms. This depends on the extent and quality of the local inflammatory response and the maintenance of a systemic inflammatory response over a long duration to disable immunoavoidance.
Based on concepts derived from actual human immune mechanisms leading to curative anti-tumour immunity and the breaking of natural immunotolerance, we propose a novel approach to cancer vaccine development, proof-of-concept having been shown in animal tumour models [6,7]. The protocol has three separate phases, the first designed to increase the circulating numbers of Th1 immune cells in cancer patients, shifting the balance from Th2 to Th1. The second elicits anti-tumour specific Th1 immunity. The third relies on activating components of the innate and adaptive immune responses to generate a sustained Th1 cytokine environment that downregulates immunoavoidance (protocol NCT00861107; www.clinicaltrial.gov).
The key component is an experimental drug, AlloStimTM, which consists of in vitro differentiated and expanded Th1 immune cells derived from normal blood donors. These cells, used in an intentionally mismatched setting, are activated at the time of injection with anti-CD3/anti-CD28 monoclonal antibody conjugated microbeads, and produce large amounts of inflammatory cytokines while expressing effector molecules on the cell surface, e.g. CD40L and FasL, that promote the Th1 immunity.
To enhance the number of circulating Th1 cells circulating in cancer patients, AlloStimTM cells will be injected intradermally each week. The alloantigens expressed on foreign cells should stimulate a potent immune rejection response. In addition, the expression of Th1 cytokines by the AlloStimTM will provide an inflammatory adjuvant environment that steers the immune response to the alloantigens on AlloStimTM cells to Th1 memory immunity. Multiple injections are expected to act as booster shots, increasing the number of circulating memory Th1 cells specific for the alloantigens.
To educate the immune system of the threat posed by the tumour, and to respond by developing tumour-specific Th1 immunity, combination of AlloStimTM with tumour cryoablation is proposed. By combining pathological tumour death8 by cryoablation with intratumoural AlloStimTM that produces inflammatory danger signals, the conditions are set for creating Th1 tumour-specific immunity. Normally cells in the body die by apoptosis, a continuous part of cell turnover. The immune system does not respond to apoptotic cells, thereby avoiding autoimmunity. Most chemotherapy agents induce tumour cell death by this same apoptotic process [9]. Cryoablation of tumour causes death by necrosis, and releases the internal contents of many cells simultaneously into the microenvironment which provide “eat me” signals to immune cells recruited into the site. According to the danger theory, DCs in the microenvironment can be programmed to mature, which promotes the development of Th1 immunity specific for the engulfed antigens.
With increased Th1 memory cells in circulation and tumourspecific CTLs, the final step is to give AlloStimTM by intravenous infusion. The intentionally mismatched allogeneic cells in immunocompetent hosts primed for the alloantigens should elicit potent rejection. This would create an immunological environment similar to the GVHD environment in allogeneic transplants [10]. However, rejection of allogeneic cells is not expected to be toxic.
CD40L (CD154) expressed on the surface of AlloStimTM should interact with CD40 constitutively expressed on host hematopoietic progenitors, epithelial and endothelial cells, and all APC, DC, activated monocytes, activated B lymphocytes, follicular DCs and NK cells. CD40L is a strong inducer of Th1 responses and its stimulation abrogates the suppressive effect of Treg cells. It also activates innate NK cells and DCs. CD40-CD40L activation of DCs leads to maturation and upregulation of co-stimulatory molecules, producing large amounts of IL-12 that has potent anti-tumour and Th1 directing properties. CD40L also has direct anti-tumour effects by suppressing growth and inducing extensive cell death [11-13]; its activation additionally enhances CTL-mediated lysis of tumours.
Th1 cytokines produced by AlloStimTM and CD40L expression also activate circulating allospecific Th1 cells created in the first phase of the protocol, thereby sustaining the necessary inflammatory environment for downregulating immunoavoidance and enabling the tumour-specific CTL created in the second phase of the protocol to mediate anti-tumour effects.
This protocol incorporating these new concepts for the development of cancer vaccines is about to be tested in the clinic The results might in due course bring us closer to realizing the promise of immunotherapy in cancer.
1. Morgan G, Ward R, Barton M. The contribution of cytotoxic chemotherapy to 5-year survival in adult malignancies. Clin Oncol (R Coll Radiol) 2004;16:549.
2. Ichim CV. Revisiting immunosurveillance and immunostimulation: Implications for cancer immunotherapy. J Transl Med 2005;3:8.
3. Prehn RT, Prehn LM. Immunostimulation of cancer versus immunosurveillance. Medicina (B Aires) 1996;56(1):65.
4. Matzinger P. Introduction to the series. Danger model of immunity. Scand J Immunol 2001;54:2.
5. Janeway CA, Jr. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol Today 1992;13:11.
6. Har-Noy M, Zeira M, Weiss L, Slavin S. Completely mismatched allogeneic CD3/CD28 cross-linked Th1 memory cells elicit anti-leukemia effects in unconditioned hosts without GVHD toxicity. Leuk Res 2008;32(12):1903.
7. Har-Noy M, Zeira M, Weiss L, Fingerut E, Or R, Slavin S. Allogeneic CD3/CD28 cross-linked Th1 memory cells provide potent adjuvant effects for active immunotherapy of leukemia/lymphoma. Leuk Res 2009;33(4):525.
8. Ablin RJ. An Appreciation and Realization of the Concept of Cryoimmunology, p. 136. In Onik, G.M.; Rubinsky, B.; Watson, G. and Ablin, R.J. (Eds.): Percutaneous Prostate Cryoablation. Quality Medical Publishing, Inc., St. Louis, 1995.
9. Hannun YA. Apoptosis and the dilemma of cancer chemotherapy. Blood 1997;89:1845.
10. Har-Noy M, Slavin, S. The Anti-Tumor Effect of Allogeneic Bone Marrow/Stem Cell Transplant Without Graft vs. Host Disease Toxicity and Without a Matched Donor Requirement? Med. Hypotheses 2008;70(6):1186.
11. Georgopoulos NT, Steele LP, Thomson MJ, Selby PJ, Southgate J, Trejdosiewicz LK. A novel mechanism of CD40-induced apoptosis of carcinoma cells involving TRAF3 and JNK/AP-1 activation. Cell Death Differ 2006;13:1789.
12. Georgopoulos NT, Merrick A, Scott N, Selby PJ, Melcher A, Trejdosiewicz LK. CD40-mediated death and cytokine secretion in colorectal cancer: A potential target for inflammatory tumour cell killing. Int J Cancer 2007.
13. Tong AW, Papayoti MH, Netto G, Armstrong DT, Ordonez G, Lawson JM, Stone MJ. Growth-inhibitory effects of CD40 ligand (CD154) and its endogenous expression in human breast cancer. Clin Cancer Res 2001;7:691.
• Richmond Prehn has discussed this problem in an earlier issue of Oncology news (Prehn RT. A cancer vaccine or just wishful thinking? Oncology News 2006;1:8-10).


